
Publicado en la colección «Derechos Intelectuales» volumen Nº17 de ASIPI
Andrés Rincón Uscátegui 
Resumen
Sin constituir activo de propiedad intelectual en sí mismo, como lo es una patente, la protección de datos de prueba que acredita seguridad y eficacia de un fármaco constituye una importante herramienta para una industria cuyos costos de investigación y desarrollo son elevados. Por esta razón, el Acuerdo sobre Aspectos de Derechos de Propiedad Intelectual Relacionados con el Comercio (ADPIC) propende por un estándar mínimo de protección para prevenir el uso comercial desleal de este tipo de información en la medida en que se trata de información no divulgada cuya generación requiere un esfuerzo considerable que de no protegerse generaría un desequilibrio de mercado basado en el desconocimiento de dicho esfuerzo. El presente artículo pretende aportar a la discusión sobre la naturaleza jurídica de protección de los datos de prueba como activo intangible, analizando en qué consiste la generación de dichos datos, cuál es el riesgo que asume quien los genera, dónde yace la no divulgación de la información, por qué no puede ser considerada información secreta, su importancia para quienes no están dispuestos a generarla, y la forma más adecuada de protegerla.
I. Introducción
Los datos de prueba hacen referencia a la generación de información necesaria para acreditar, ante una autoridad sanitaria de un país determinado, la seguridad y eficacia de un producto cuya comercialización está regulada por el Estado por razones de salud pública. El tema ha tenido su principal desarrollo en los sectores de agroquímicos y farmacéuticos, pues son sectores donde la industria de investigación y desarrollo tiene que invertir sorprendentes cantidades de recursos económicos y humanos durante largos periodos de tiempo, para obtener la información que finalmente permite acreditar esa seguridad y eficacia requeridas para garantizar que el producto puede ser finalmente consumido directa o indirectamente por los seres humanos. La necesidad de implementar sistemas de protección específicos para impedir el uso comercial desleal de esta información ha sido particularmente controvertida en la industria farmacéutica, dadas las implicaciones que esto puede traer desde el punto de vista de acceso a medicamentos.
Aunque el tema de datos de prueba como tal no hace referencia a un derecho específico de propiedad industrial, ya que a lo sumo el dossier farmacológico de un producto farmacéutico en sí mismo constituiría una recopilación de información amparada por el derecho de autor como una tediosa obra de carácter científico, es común que la generación de dicha información tienda a asociarse con dos activos de propiedad industrial: (i) las patentes de invención, ya que bien puede darse el caso de que el principio activo que compone el medicamento cuente con protección de patente sobre la molécula como tal, sobre una formulación específica de ella, o sobre alguna de sus sales o ésteres; y (ii) los secretos empresariales, ya que en cualquier caso un dossier farmacológico adecuadamente preparado derivaría de información que no ha sido previamente divulgada, que ni siquiera se encuentra incluida dentro del dossier, que es particularmente referida a los pacientes sobre quienes fueron practicados las pruebas de seguridad y eficacia correspondientes y que, en últimas, es la que permite validar cualquier conclusión de seguridad y eficacia arrojada por el estudio.
Si bien la protección de datos de prueba como tal no se puede asimilar a una protección de patente o de secreto empresarial, por el hecho de involucrar un porcentaje de información no divulgada, sí amerita una protección especial e integral, ya que el uso indiscriminado por parte de terceros de la fracción divulgada de la información puede tener efectos competitivos desleales en un mercado determinado. En el caso concreto, en materia farmacéutica, el hecho de permitir que un tercero se apoye en información divulgada sin tener que generar la fracción de información no divulgada, que es necesaria para validarla, implica en últimas un desequilibrio de mercado en favor del tercero y en detrimento del innovador. Es por ello que la información de datos de prueba correspondiente a nuevas entidades químicas fue amparada en materia regulatoria en el Acuerdo sobre Aspectos de Propiedad Intelectual relacionados con el Comercio (ADPIC) (1994), con protección sobre la información no divulgada contra todo uso comercial desleal.
Los Miembros, cuando exijan como condición para aprobar la comercialización de productos farmacéuticos o de productos químicos agrícolas que utilizan nuevas entidades químicas, la presentación de datos de pruebas u otros no divulgados cuya elaboración suponga un esfuerzo considerable, protegerán esos datos contra todo uso comercial desleal. Además, los Miembros protegerán esos datos contra toda divulgación, excepto cuando sea necesario para proteger al público, o salvo que se adopten medidas para garantizar la protección de los datos contra todo uso comercial desleal [1].
La inclusión en el ADPIC de dicha protección, aunque probablemente necesaria para generar la obligación de implementación de disposiciones nacionales con estándares mínimos que en efecto protegiesen el uso comercial desleal de los datos de prueba, fue cuando menos desafortunada en su redacción, ya que (i) parte de un supuesto absoluto, no aplicable por lo general a los datos de prueba, cual es el de la falta de divulgación de la información, sin aclarar qué pasa con el porcentaje de la información por lo general divulgado; y (ii) limita la información que debe protegerse a aquella cuya generación hubiese implicado la realización de un esfuerzo considerable, como si acaso hubiese casos en donde dicho esfuerzo pudiese ser inexistente.
El problema se acrecienta si se tiene en cuenta que el ADPIC hace parte de un tratado multilateral que no es de aplicación inmediata y que requiere por parte de los países Miembros de la OMC un esfuerzo de implementación de normas con las cuales se cumple con los estándares mínimos del tratado y se hace uso de sus flexibilidades.
El uso comercial desleal de los datos de prueba debe prevenirse garantizando un derecho sui generis en las legislaciones internas de los Miembros de la OMC, sin que su alcance esté limitado al hecho de haber sido o no previamente divulgados (ya que normalmente en su parte no confidencial deben haber sido previamente debatidos), sino por el esfuerzo que implica la generación de su parte no divulgada en términos económicos y de recurso humano, esfuerzo que se presume cuando el resultado es la demostración de la seguridad y eficacia de un producto determinado, expresada en un dossier farmacológico.
II. ¿Qué son los datos de prueba de productos farmacéuticos y por qué su generación amerita protección?
Existen millones de compuestos que día a día son analizados en los laboratorios de empresas farmacéuticas de investigación y desarrollo, donde eventualmente un número muy reducido presenta características de interés biológico y amerita pasar a una posterior etapa de evaluación farmacológica y de seguridad preclínica, que se lleva a cabo en animales y modelos computacionales. De estos compuestos, unos pocos califican para pasar a evaluación en humanos, donde inician los ensayos clínicos y se generan los datos de prueba. Esta etapa de investigación, previa a los ensayos clínicos en humanos, fácilmente puede tardarse en promedio cinco años.
Los ensayos clínicos con los que se generan los datos de prueba se dividen en tres fases sucesivas: la fase I, con la que se pretende demostrar que el producto es tolerable; la fase II, que busca determinar una dosis óptima; y la fase III, que es la que finalmente arroja evidencia de la seguridad y eficacia de la nueva entidad química en humanos.
A lo largo de esas fases, que pueden demorarse entre cinco a diez años adicionales a los cinco de la fase preclínica, se desarrollan cientos de estudios de los cuales se deriva información de miles de pacientes. La información de pacientes siempre es confidencial por razones de privacidad[2] y los costos generados en esta fase de investigación y desarrollo son mayormente asumidos por el laboratorio innovador del producto.
Tan solo un puñado de compuestos de todos los seleccionados para la realización de ensayos clínicos llega a una fase de aprobación ante una autoridad regulatoria y de esos tan solo uno o dos llegan finalmente a comercializarse, lo que hace de la inversión en tiempo y dinero un riesgo altísimo asumido por el innovador. No hay ninguna seguridad de éxito.
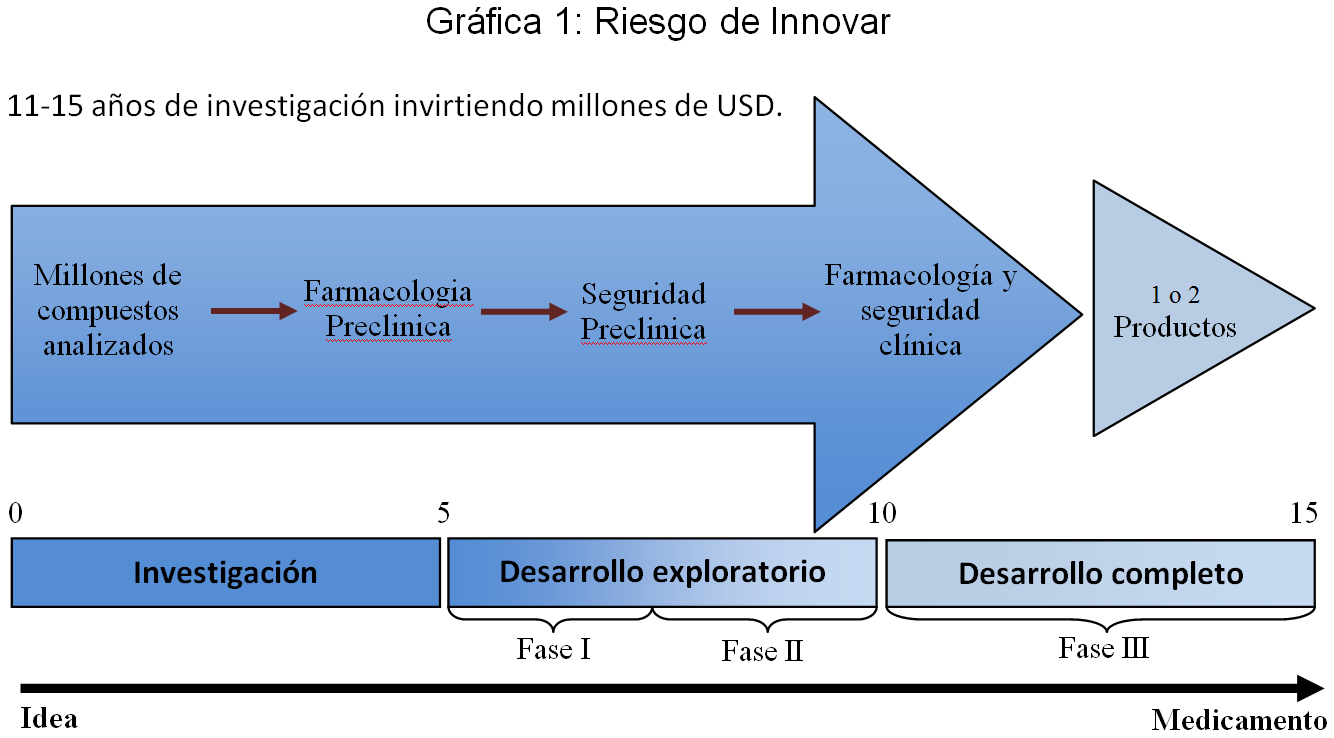
Lo que debe garantizar cualquier legislación nacional para brindar una protección efectiva contra el uso comercial desleal por parte de terceros es que se impida que estos, que no intervinieron en el proceso de investigación y desarrollo, se aprovechen gratuitamente del esfuerzo considerable que naturalmente implica la realización de ensayos clínicos; esfuerzo reflejado a lo largo del tiempo en recursos humanos y millonarios recursos financieros[3].
Sin una protección adecuadamente establecida, el aprovechamiento ocurre al momento en que un genérico solicita un registro sanitario aportando datos públicamente disponibles de seguridad y eficacia del producto que no ha tenido que generar, ni tiene cómo validar. Mientras el innovador debe invertir y arriesgar mucho tiempo y recursos financieros en la determinación de seguridad y eficacia de la nueva entidad química, el solicitante genérico, que no ha invertido un solo centavo en tal determinación, simplemente espera a que el innovador haya demostrado seguridad y eficacia para solicitar la aprobación de su producto sin tener que generar sus propios datos de prueba.
Al existir un sistema de protección adecuado, cualquiera que este sea, lo que se debe lograr es nivelar la barrera de entrada al mercado que tiene el innovador con respecto a sus copiadores, debiendo ambos generar los datos correspondientes o, de no ser así por parte del copiador, sufrir algún tipo de limitación que compense o impida su acceso de manera provisional o permanente en un mercado determinado.
III. ¿Cuál es el sistema de protección ideal?
Además de las dificultades que ya ofrece la desafortunada redacción del artículo 39.3 del ADPIC, en la que se sugiere un estándar de protección limitado a información no divulgada que haya representado un esfuerzo considerable, se le une el hecho de que las expresiones “nueva entidad química”, “información no divulgada” y “esfuerzo considerable” no fueron definidas dentro del estándar, como tampoco lo fue la expresión “uso comercial desleal”. Esta falta de precisión en el alcance del artículo hace que su implementación por parte de los países de la OMC sea tan variada y diversa como el número de miembros que componen esta organización. No obstante, dentro de esa variedad de sistemas se identifican dos tendencias primordiales para procurar evitar el uso comercial desleal de la información en materia regulatoria: (i) aquellas que sugieren, como en Estados Unidos[4] y Europa[5], que el uso comercial desleal se evita garantizando periodos de protección limitados en el tiempo (de entre 5 a 10 años), dentro de los cuales ningún tercero podrá solicitar autorización de comercialización a menos de que acredite la realización de estudios propios de seguridad y eficacia; y (ii) aquellos que consideran que será posible solicitar autorización de comercialización siempre y cuando la información haya sido accedida de una manera honesta (propuesta primordialmente encabezada por países miembros del Grupo Africano)[6].
En el contexto latinoamericano, aunque resulta difícil en el marco de este documento hacer un análisis pormenorizado de la legislación de cada país, se pueden encontrar ejemplos claros de las dos tendencias mencionadas. Basta analizar al interior de los países miembros de la Comunidad Andina para darse cuenta de que frente a una misma norma de implementación –el Régimen Andino de Propiedad Industrial[7]– coexisten ambas posturas. Hay sin embargo que aclarar que esta disposición no hizo nada distinto que replicar la disposición del artículo 39.3 del ADPIC, reiterando que los países miembros podrían tomar las medidas que consideraran pertinentes para garantizar la protección contra el uso comercial desleal de los datos de prueba. La disposición tiene un agravante desde el punto de vista de su interpretación: se encuentra incluida dentro del título XVI, referido a la competencia desleal vinculada a la propiedad industrial, pero en su capítulo II, referido a los secretos empresariales.
Los Países Miembros, cuando exijan como condición para aprobar la comercialización de productos farmacéuticos o de productos químicos agrícolas que utilizan nuevas entidades químicas, la presentación de datos de pruebas u otros no divulgados cuya elaboración suponga un esfuerzo considerable, protegerán esos datos contra todo uso comercial desleal. Además, los Países Miembros protegerán esos datos contra toda divulgación, excepto cuando sea necesario para proteger al público, o salvo que se adopten medidas para garantizar la protección de los datos, contra todo uso comercial desleal. Los Países Miembros podrán tomar las medidas necesarias para garantizar la protección consagrada en este artículo [8].
A. Colombia
Siendo pionero en la materia al interior de la Comunidad Andina, y generando bastante controversia, el gobierno colombiano optó por implementar un sistema de protección para la información no divulgada de nuevas entidades químicas[9] por un periodo de 5 años, dentro de los cuales ningún tercero que no acreditase la realización de estudios propios de seguridad y eficacia podría solicitar un registro sanitario sobre el mismo medicamento[10].
Esta determinación generó una enorme controversia en cuanto a la posibilidad de los países miembros de la Comunidad Andina para establecer periodos de protección, llegándose incluso en un primer momento a considerar que la adopción de este tipo de disposiciones constituía una violación de la Decisión 486 de 2000, que no había contemplado expresamente dicha posibilidad[11]. La controversia fue finalmente dirimida dentro de la misma Comunidad Andina, que, mediante el órgano competente, la Comisión de la Comunidad Andina, tuvo que emitir una decisión interpretativa del artículo 266 de la Decisión 486 para determinar que en efecto dicho artículo debía interpretarse en el sentido de permitir la adopción por parte de los países miembros de periodos de exclusividad[12]. La determinación fue sin duda uno de los catalizadores que generó la salida de Venezuela de la Comunidad Andina.
Posteriormente, Colombia suscribió tratados de libre comercio (TLC) con los Estados Unidos, la Unión Europea y EFTA, en los cuales se incluyeron disposiciones tendientes a garantizar datos de prueba por periodos de tiempo de cuando menos 5 años, estándar con el que ya se cumplía internamente[13].
Desde su implementación en 2002, más de 160 moléculas han sido amparadas con protección de datos. En la actualidad más de 100 moléculas cuentan con protección vigente y alrededor de 20 están buscando ser protegidas[14]. La protección ha sido efectiva y no existen récords de compañías genéricas que hayan solicitado autorización de comercialización acreditando estudios propios. Sin embargo, tanto el Decreto 2085 como las resoluciones que conceden protección de datos son objeto de constantes ataques legales tanto internamente como en el ámbito andino[15].
B. Ecuador
En el Ecuador, por el contrario, se optó por no garantizar un periodo limitado de protección; en su lugar se decidió contemplar como un acto de competencia desleal “el uso comercial desleal de datos de pruebas no divulgadas u otros secretos cuya elaboración suponga un esfuerzo considerable…”[16], lo que en últimas supone una aplicación del sistema según el cual los datos pueden ser aprovechados siempre y cuando la información haya sido accedida de una manera honesta. Lo anterior con un agravante: la norma ecuatoriana parte del supuesto de que los datos de prueba son verdaderos secretos empresariales, lo que hace que en la práctica la protección sea inexistente[17].
C. Perú
Perú, por su parte, y en cumplimiento de obligaciones adquiridas en el marco del TLC suscrito con Estados Unidos en 2006, implementó al igual que Colombia un sistema de protección basado en límites de tiempo, en este caso también de 5 años para nuevas entidades químicas[18].
D. Bolivia y Venezuela
Hasta 2010 Bolivia tuvo vigente un TLC suscrito con México desde 1995 de acuerdo con el cual se encontraba obligada a proveer un periodo de protección razonable a los datos de prueba, sin que el documento especificara cuál debería ser el término de dicho periodo. Pese a la existencia de esta obligación, nunca existió en 16 años de vigencia una disposición legal en el país que diera cumplimiento al tratado[19], que finalmente fue reemplazado por un Acuerdo de Complementación Económica entre ambas naciones, limitado a aspectos comerciales y manteniendo condiciones arancelarias, pero sin incluir disposición alguna en materia de protección de datos. Por su parte, Venezuela, aunque ya no es miembro de la Comunidad Andina, no ha desarrollado regulación de algún tipo en este sentido.
IV. ¿Qué se debe entender por “información que no haya sido previamente divulgada”?
Todo dossier farmacológico elaborado por una compañía de investigación y desarrollo para demostrar la seguridad y eficacia de una nueva entidad química que aspira comercializar en un mercado determinado está elaborado con base en innumerables estudios de los cuales se han recolectado datos absolutamente confidenciales.
De esos datos originarios, primordialmente determinados por la información recaudada de pacientes, se generan una serie de análisis, reportes y conclusiones iniciales que no están disponibles al público, con base en los cuales se generan estudios y artículos científicos derivados de la información confidencial recolectada. Estos estudios y artículos se ponen a disposición del público especializado y son en efecto públicos, ya que una vez obtenidos y analizados los datos del paciente es fundamental que sean debatidos y controvertidos por la comunidad científica en general, cuyo análisis y discusiones especializadas contribuyen a evitar sesgos en quien genera la información y dan a conocer los adelantos e inconvenientes encontrados entre la comunidad médica que tendrá acceso al producto si este finalmente sale al mercado. Sin embargo, en ningún caso es el objetivo de esta divulgación permitir a otros el uso de esta información, en especial por el esfuerzo y riesgo asumido para generarla.
Un dossier farmacológico que se presenta a una autoridad regulatoria normalmente se compone de información derivada, mayormente divulgada, pero cuya validación solo se puede obtener con acceso a la información originaria que, como ya se dijo, es de carácter confidencial.
Apoyado en la metáfora del témpano, utilizada comúnmente para ilustrar este aspecto, la información divulgada y la no divulgada constituyen un conjunto indivisible para efectos de su protección; es solamente con todo el conjunto que se puede demostrar y validar la seguridad y eficacia de una nueva entidad química. Negar la protección por el hecho de que parte de la información no es confidencial equivaldría entonces a negar la existencia y justificación de la protección de datos en sí misma, ya que, en ningún caso, el dossier farmacológico que respalda la solicitud de protección está apoyado en información cien por ciento no divulgada.
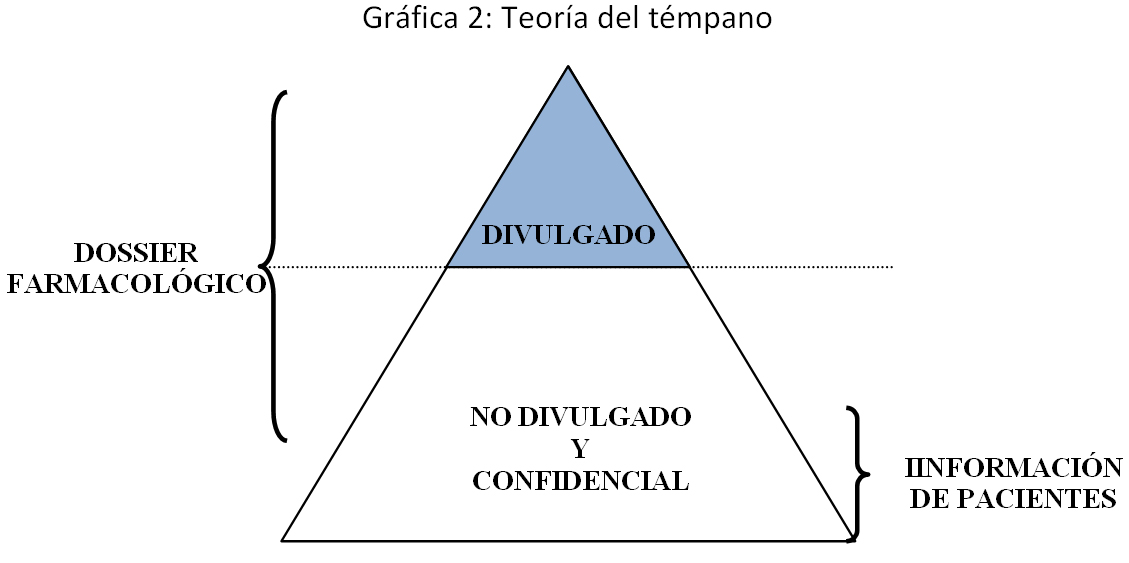
De acuerdo con esta interpretación, una protección efectiva no debe solo extenderse a los datos de prueba no divulgados que reposan físicamente en el dossier, sino a todos aquellos datos que lo componen, ya que de solo protegerse aquello nunca antes divulgado incluido en el dossier, se dejaría por fuera de protección la porción divulgada de la información, porción que únicamente puede ser validada con los datos no divulgados de los cuales esta deriva y en los cuales se respalda. Una posición contraria que requiera la confidencialidad de toda la información relacionada con la seguridad y eficacia de un medicamento para que se pueda dar una protección de datos, o que solo proteja aquello que se demuestre que nunca antes ha sido divulgado, sería contraproducente para la salud pública, pues obligaría a las compañías farmacéuticas de investigación y desarrollo a esconder sus resultados, limitando así una importantísima labor de controversia científica que actualmente se da gracias a la publicación de resúmenes y artículos de tales estudios clínicos.
V. ¿Qué se debe entender por “esfuerzo considerable”?
El esfuerzo considerable es un concepto que, en materia de protección de datos de prueba, no se encuentra definido formalmente en un texto legal internacional, regional o local. Y no lo está primordialmente porque el esfuerzo considerable es inherente a la generación de datos de prueba.
Es claro que para lograr demostrar seguridad y eficacia de una nueva entidad química y obtener aprobación para su comercialización es necesario, con posterioridad a una fase preclínica en la que ya se invirtieron un tiempo de en promedio cinco años y numerosos recursos financieros, realizar ensayos clínicos compuestos por una numerosa cantidad de exámenes practicados en humanos. Esos ensayos clínicos, además de demorar un promedio de diez años en su adecuada realización, requieren cada vez de inversiones más altas de dinero, puesto que cada día existen nuevos requerimientos sanitarios que la industria de investigación y desarrollo debe cumplir.
Pretender probar un esfuerzo considerable en materia de datos de prueba con algo más que la presentación de los estudios mismos equivale a presumir que los ensayos clínicos no fueron practicados, ya que no puede haber desarrollo de nuevas entidades químicas utilizables en humanos, como productos farmacéuticos, sin la realización de un esfuerzo considerable en tiempo, capacidad operativa y recursos financieros. Por eso es común encontrar sistemas en los cuales la realización del esfuerzo considerable, en mala hora requerido por ADPIC, simplemente se presume o se acredita con una declaración jurada[20].
Finalmente, y aun acudiendo simplemente al factor económico para determinar si existe o no un esfuerzo considerable, es claro que el costo promedio para desarrollar los ensayos clínicos de una nueva entidad química es de aproximadamente ¡USD 467 millones![21]. Aun, si se considera en gracia de discusión que esta cifra no es exacta, pero que se trata en realidad de la mitad, o de una cuarta parte, se está hablando como mínimo de más de USD 100 millones, cuya inversión constituye sin duda un esfuerzo tan considerable que una industria genérica[22] no está dispuesta a incurrir en él para participar en el mercado. En últimas, el criterio de cuánto constituye o no un esfuerzo considerable se vuelve más un tema de umbral probatorio necesario para optar a la protección, umbral que pese a lo obvio del esfuerzo, como se ha resaltado, es discrecional de la autoridad nacional[23].
VI. ¿Qué diferencia tiene la protección sobre los datos de prueba con respecto a la protección que se obtiene con una patente?
Debido a la vinculación que existe entre industria farmacéutica y propiedad industrial, y particularmente entre esta industria y el derecho de patentes, es común que quienes critican los mecanismos de protección de datos de prueba tiendan a asociar estos sistemas de protección con mecanismos de extensión de derechos de patente, o incluso a equiparar los datos de prueba con pequeñas patentes de invención.
Esta asociación es incorrecta, ya que el producto que pretende comercializarse y cuya autorización sanitaria es solicitada en un determinado país puede o no contar con protección de patente, e incluso el hecho de corresponder a una molécula patentada no es siquiera un indicativo de cumplimiento de requisitos legales para acceder a protección de datos de prueba. Bien puede darse el caso de moléculas patentadas que incluyan entidades químicas que no puedan ser consideradas como nuevas a la luz del derecho regulatorio, pese al hecho de que en su momento resultaron novedosas e inventivas para efectos de la solicitud de patente en un territorio determinado.
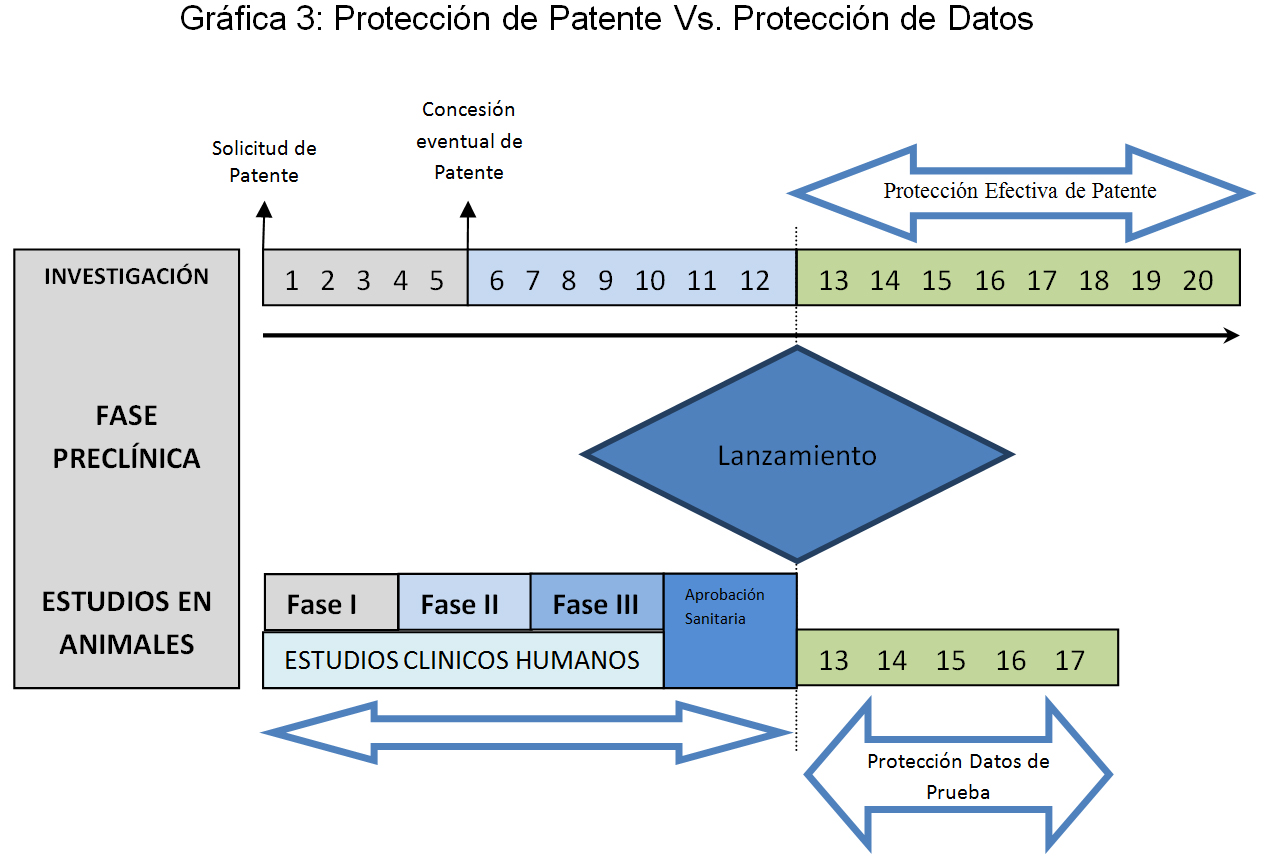
Por definición, el derecho de patente confiere a su titular la atribución de impedir a terceras personas que no tengan su consentimiento la reproducción con fines de mercado del producto o el procedimiento finalmente reivindicado y concedido en el título de patente. Muy distinto a esto, lo que protege el derecho sobre datos de prueba es su uso comercial desleal de los datos generados por otro, mas no se impide la realización de estudios propios con los que se pueda comprobar bajo estándares internacionales que un producto determinado, patentado o no, es seguro y eficaz.
Por último, asumiendo que el producto sobre el cual se acredita seguridad y eficacia coincide con alguna reivindicación preferida de una solicitud de patente, el término efectivo de protección sobre los datos de prueba a lo sumo será concomitante con el de expiración de protección sobre la patente concedida, pero en ningún caso superior, por lo cual es improbable que alguien pensare en utilizar el sistema para extender artificialmente el periodo de protección de la patente correspondiente, máxime considerando que su competidor está en libertad, y hoy en día, sin dudarlo, en la capacidad de generar estudios propios con los cuales podría entrar al mercado una vez expirada la patente.
VII. ¿Por qué no se trata de un secreto empresarial?
Si los datos de prueba fuesen fuente de secretos empresariales, los laboratorios de investigación y desarrollo no estarían dispuestos a divulgarlos sin contar para ello con protección de patente o un derecho equivalente que les garantizara que nadie más pueda replicarlos, ya que su valor se derivaría del hecho de ser secretos y no del hecho de demostrar seguridad y eficacia. Lo anterior, naturalmente es indeseable, ya que entre otras implicaría que los datos de prueba contarían con una especie de protección eterna en tanto permanecieran secretos, e independientemente del paso del tiempo, la única forma de apoyarse en ellos sería replicando los propios. Esto generaría un retardo indefinido en la entrada de productos equivalentes en términos de seguridad y eficacia, lo cual iría en detrimento de la sociedad y de la salud pública.
Si bien es cierto que los datos de prueba siempre derivan de información altamente confidencial de pacientes, también es cierto que en su mayoría se componen de información que ya ha sido previamente revelada de manera voluntaria, particularmente entre la comunidad científica, con el objeto de garantizar su controversia por pares académicos, lo que entre otras ayudará a minimizar el riesgo de solapamiento de errores o imprecisiones en la apreciación de resultados.
VIII. Conclusiones
El ADPIC impuso un estándar internacional que incluye el deber de provisión de protecciones específicas para prevenir el uso comercial desleal de información no divulgada que demuestre la seguridad y eficacia de una nueva entidad química.
Lamentablemente, el estándar ADPIC no definió lo que ha de entenderse por “nueva entidad química”, “información no divulgada”, “esfuerzo considerable” o “uso comercial desleal”, lo que deja abierta la puerta a tantas formas de regulación de la materia como países miembros de la OMC.
Las tendencias legislativas más efectivas son aquellas que imponen la existencia de periodos de tiempo de entre 5 a 10 años, dentro de los cuales cualquier tercero interesado en comercializar un producto que contenga la nueva entidad química debe acreditar la realización de estudios propios que demuestren su seguridad y eficacia.
Los países que no imponen la existencia de periodos de tiempo son en la práctica ineficaces y permiten que terceros que no han generado datos propios soliciten autorización de comercialización una vez el innovador ha acreditado seguridad y eficacia, basándose para ello en información derivada que se encuentra disponible, y que normalmente ha sido puesta a disposición de la comunidad científica por el mismo innovador.
Si bien es cierto que la información sobre la seguridad y eficacia de un producto farmacéutico puede encontrarse parcialmente disponible, como por ejemplo en Internet, también es cierto que dicha información es derivada y constituye resúmenes y resultados de investigaciones mucho mas amplias originadas en el recaudo y análisis de información que no se encuentra divulgada, y que en el más estricto sentido nunca será divulgada por su naturaleza confidencial.
La información de pacientes de los cuales se obtienen los resultados que son finalmente debatidos y publicados siempre será confidencial, al punto que solo será revelada a una autoridad competente para efectos de validación de resultados específicos.
La única forma que existe de validar información derivada de seguridad y eficacia de medicamentos es teniendo acceso a información no divulgada y confidencial de pacientes, que solo posee quien en efecto realiza estudios de fase I, fase II y fase III. Es por ello que, sin entrar a discriminar entre información no divulgada y divulgada, la información de datos de prueba debe protegerse como un todo indivisible.
No existen datos de prueba generados sin incurrir en un esfuerzo considerable medido en tiempo, recurso humano, científico y económico. Por tanto, el esfuerzo considerable debe presumirse de quien aporta un dossier farmacológico completo, cuya información se encuentra en capacidad de validar con la información confidencial, a la que se tiene acceso por el hecho de haber realizado los estudios correspondientes.
De presentarse información derivada (y en principio no protegida) de seguridad y eficacia a efectos de solicitar autorización de comercialización de un producto farmacéutico, dicha autorización debe ser negada hasta que se demuestre que quien la aporta tiene cómo validarla.
La protección otorgada al generador de información vía datos de prueba es una protección sui generis que no coincide con derecho alguno de propiedad industrial y que no se asimila a la protección que otorga una patente.
Por último, la protección otorgada al generador de información vía datos de prueba no debe otorgarse bajo el supuesto de que deba demostrarse que es secreta, ya que lo que verdaderamente constituye un secreto empresarial, que es la información de pacientes, es una herramienta de validación que normalmente ni siquiera se incluye en el dossier farmacológico.
BIBLIOGRAFÍA
Doctrina
Basheer, Shamnad, Protection of Regulatory Data Under Article 39.3 of TRIPS: The Indian Context, Intellectual Property Institute (IPI), disponible en SSRN: http://ssrn.com/abstract=934269
Joseph A DiMasi et ál., The Price of Innovation: new estimates of drug development costs, publicado por el Journal of Health Economics, 2003, 22, pp. 151-185. Disponible en la web: http://moglen.law.columbia.edu/twiki/pub/LawNetSoc/BahradSokhansanjFirstPaper/22JHealthEcon151_drug_development_costs_2003.pdf.
TRIPS Council, African Group et al, 2001 disponible en la web: http://www.who.int/intellectualproperty/topics/ip/tHoen.pdf
Normatividad
Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC) disponible en la web: http://www.wto.org/spanish/docs_s/legal_s/27-trips.pdf
Comunidad Andina, Comisión de la comunidad Andina, Decisión 486 de 2000, disponible en la web: http://intranet.comunidadandina.org/Documentos/Gacetas/Gace2146.pdf
Comunidad Andina, Comisión de la comunidad Andina, Decisión Andina 632 de 2006, disponible en la web: http://www.wipo.int/wipolex/es/text.jsp?file_id=223759
Declaración de Helsinki, Principios Éticos para Investigaciones Médicas en Seres Humanos Disponible en la web: http://www.wma.net/es/30publications/10policies/b3/17c_es.pdf
Decreto 2085 de 2002 disponible en la web: http://www.presidencia.gov.co/prensa_new/decretoslinea/2002/septiembre/19/dec2085190902.pdf
Ecuador, Codificación de la Ley de Propiedad Intelectual N° 426 de 2006 disponible en la web: http://www.wipo.int/wipolex/en/text.jsp?file_id=195678
Perú: Decreto Legislativo 1072 de 2008 y reglamentado mediante Decreto Supremo 002-09-SA disponible en la web: http://www.wipo.int/wipolex/es/text.jsp?file_id=185065
TLC con Estados Unidos disponible en la web: http://www.ustr.gov/webfm_send/1336
TLC con la Unión Europea disponible en la web: http://trade.ec.europa.eu/doclib/docs/2011/march/tradoc_147704.pdf
TLC con EFTA disponible en la web: http://www.efta.int/~/media/Documents/legal-texts/free-trade-relations/columbia/EFTA-Colombia%20Free%20Trade%20Agreement%20EN.pdf
TLC Bolivia – México disponible en la web: http://www.sice.oas.org/TPD/BOL_MEX/BOL_MEX_s.ASP
Jurisprudencia
Comisión Europea, proceso EC No. 3286/94, Federación Europea de Industria y Asociaciones (EFPIA) contra Turquía disponible en la web: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C:2003:311:0031:0032:ES:PDF
Comunidad Andina, Tribunal de Justicia de la Comunidad Andina, Proceso 194-AI-2004, disponible en la web: http://intranet.comunidadandina.org/documentos/Gacetas/Gace1295.pdf Comunidad Andina, Tribunal de Justicia de la Comunidad Andina, Proceso 02-AI-2009, disponible en la web: http://intranet.comunidadandina.org/documentos/Gacetas/Gace1987.pdf





